Michael Fischer.
Physical Chemistry Chemical Physics (2015) 17, 25260-25271.
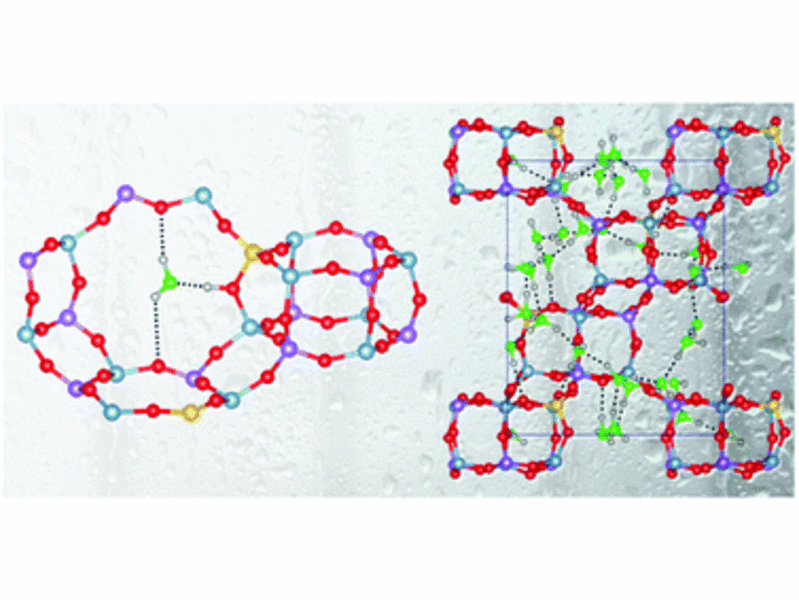
The chabazite-type silicoaluminophosphate SAPO-34 is a promising adsorbent for applications in thermal energy storage using water adsorption–desorption cycles. In order to develop a microscopic understanding of the impact of local heterogeneities and defects on the water adsorption properties, the interaction of different models of SAPO-34 with water was studied using dispersion-corrected density-functional theory (DFT-D) calculations. In addition to SAPO-34 with isolated silicon atoms, the calculations considered models incorporating two types of heterogeneities (silicon islands, aluminosilicate domains), and two defect-containing (partially and fully desilicated) systems. DFT-D optimisations were performed for systems with small amounts of adsorbed water, in which all H2O molecules can interact with framework protons, and systems with large amounts of adsorbed water (30 H2O molecules per unit cell). At low loadings, the host–guest interaction energy calculated for SAPO-34 with isolated Si atoms amounts to approximately −90 kJ mol−1. While the presence of local heterogeneities leads to the creation of some adsorption sites that are energetically slightly more favourable, the interaction strength is drastically reduced in systems with defects. At high water loadings, energies in the range of −70 kJ mol−1 are obtained for all models. The DFT-D interaction energies are in good agreement with experimentally measured heats of water adsorption. A detailed analysis of the equilibrium structures was used to gain insights into the binding modes at low coverages, and to assess the extent of framework deprotonation and changes in the coordination environment of aluminium atoms at high water loadings.